Vol. 2 (2022), Article ID 246146, 6 pages
Methodology Report
Generating a New sgRNA Vector, pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR, to Improve Base Editing
Tolulope E. Ayo and Hao Xu
Center for Molecular and Cellular Biosciences, School of Biological, Environmental, and Earth Sciences, University of Southern Mississippi, Hattiesburg, MS 39406, USA
Received 4 July 2022; Accepted 3 November 2022; Published 17 December 2022
Tolulope E. Ayo and Hao Xu, Generating a New sgRNA Vector, pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR, to Improve Base Editing, Journal of Genome Editing and Regulation, 2 (2022), art246146. doi:10.32371/jger/246146
CRISPR/Cas9-mediated base editing introduces point mutations in cellular DNA by exploiting target-specific single guide RNA (sgRNA) along with a genetically modified Cas9. Existing plasmid vectors for sgRNA expression in base editing contain either a fluorescent marker or an antibiotic resistance cassette but not both, preventing simultaneous monitoring and enrichment of transfected host cells. In this study, we introduced a fluorescent marker into pGL3-U6-sgRNA-PGK-puromycin, a popular sgRNA expression vector available at Addgene. Specifically, the cDNAs of mRFP and a T2A linker were inserted in between the hPGK promoter and the puromycin resistance gene (PuroR). After correct insertion was verified by DNA sequencing, this new plasmid, pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR, was utilized to generate a stop codon in the second exon of the Munc13-1 gene in RBL-2H3 cells. Both the mRFP fluorescent marker and the puromycin resistance marker functioned accordingly in the process. This new sgRNA vector therefore represents a useful addition to the CRISPR tool kit.
CRISPR/Cas9; base editing; mRFP; puromycin; RBL-2H3; Munc13-1
The classic form of CRISPR/Cas9 genome editing relies on RNA-guided Cas9 endonuclease to generate site-specific double-strand breaks (DSBs) in host chromosomal DNAs [1,2]. Deletion or insertion mutations are then introduced when the host cells attempt to repair these DSBs, via either the non-homologous end joining pathway or the homology directed repair pathway [3,4]. However, the induction of DSBs by the wildtype Cas9 often triggers cell death, as the repair pathways involved are not very efficient [5,6,7]. An alternative approach, known as CRISPR/Cas9 base editing, exploits instead Cas9 mutants such as nCas9 (Cas9 nickase) or dCas9 (catalytically dead Cas9) [8,9] that are attached to a base editor (e.g., cytidine deaminase). By converting C to T (G to A in the complementary strand) in selected glutamine (CAA, CAG), arginine (CGA), or tryptophan (TGG) codons in the early exons of a target gene, CRISPR/Cas9 base editing can create STOP codon mutations that prematurely terminate translation [10,11].
An indispensable tool in CRISPR/Cas9-mediated base editing is the base editing vector that expresses cytidine deaminase (APOBEC enzyme), dCas9 or nCas9, and uracil glycosylase inhibitor. There have been several generations of the cytosine base editor with increasing base editing efficiency [8,9,12]. Another critical tool is the sgRNA vector, in which target-specific sgRNAs are cloned. While the base editing vectors in general lack any markers, the sgRNA vectors available for base editing contain either an antibiotic resistance marker or a fluorescent marker, but not both [13,14]. This has prevented simultaneous monitoring and enrichment of transfected host cells, particularly in laboratories that do not have access to a fluorescence-activated cell sorter (FACS). It sometimes forces researchers to co-transfect host cells with a third plasmid (with the desired, additional marker) [12], which could increase the risk of cell toxicity.
To overcome this limitation, we set out to build a more versatile sgRNA vector that carries both fluorescent and antibiotic resistance markers. By homology-based cloning, we inserted mRFP into the widely-used pGL3-U6-sgRNA-PGK-puromycin plasmid [14] and provided evidence that both the RFP fluorescent marker and the puromycin resistant marker in the new plasmid were functional. Furthermore, this new construct was shown to be highly effective in introducing early stop codons in the Munc13-1 gene in RBL-2H3 cells.
Unless otherwise specified, DNA primers (Table 1) were designed using ApE (A plasmid Editor; by M.Wayne Davis) and custom made (Sigma, USA). Tm of the primers were determined using NEB calculator. MCS1-EF1α-RFP-T2A-Puro-pA-MCS2 was purchased from System Biosciences (#HR110PA-1). pGL3-U6-sgRNA-PGK-puromycin was a gift from Xingxu Huang (Addgene plasmid #51133), whereas pCMV_BE4max was a gift from David Liu (Addgene plasmid #112093) [12,14]. Plasmids were isolated using QiAprep Spin Miniprep kit (Qiagen, Germany) by following the manufacturer’s protocol. Plasmid DNA was quantified using NanoDrop One (Thermo Scientific, USA). DNA sequencing analysis was conducted at Primordium Labs and Eurofins.

Table 1: Oligos used in this study.
The methylated pGL3-U6-sgRNA-PGK-puromycin vector DNA was isolated from DH5α and linearized at position 1168/9 via PCR. PCR (Table 2) was conducted in a 50 μL reaction that included 50 ng of vector DNA, forward and reverse primers (VectorF and VectorR) at 500 nM each, 200 μM dNTPs, 10 μL of 5× Q5 reaction buffer, and 2 units of Q5 hot-start DNA polymerase (NEB, USA). After the PCR, 45 μL of the PCR product were mixed with 6 μL of 10× cut-smart buffer and 20 units of DpnI (NEB) in a total volume of 60 μL. Following incubation at 37 °C for 1 h—to degrade the non-linearized, template DNA that were marked by methylation—the sample was heat shocked at 80 °C for 20 min to inactivate DpnI. The linearized vector that remained was then purified using QIAquick PCR purification kit (Qiagen) by following the manufacturer’s instructions.

Table 2: PCR parameters for linearizing the pGL3-U6-sgRNA-PGK-puroR vector.
From plasmid MCS1-EF1α-RFP-T2A-Puro-pA-MCS2, the mRFP cDNA and the downstream T2A linker were amplified in a 50 μL touchdown PCR (Table 3) that contains 150 ng of the template DNA, 200 μM dNTPs, 5 μL of 10× pfu buffer, 5 units of Phusion DNA polymerase (G-Biosciences), and 200 nM forward and reverse primers (mRFPF and mRFPR). Each of the two primers has a 5′ overhang that overlaps with a corresponding sequence of the linearized vector (Table 1; colored). The insert (mRFP-T2A) including the overhangs was gel purified using QIAquick gel extraction kit (Qiagen) by following the manufacturer’s instructions.

Table 3: Touchdown PCR parameters for amplifying the mRFP-T2A insert.
After cold fusion reaction and transformation, individual bacterial colonies (that might carry pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR) from LB/ampicillin plates were suspended in 20 μL cracking buffer (1 M NaOH, 10% SDS, 5 mM EDTA) for 10 min at 68 °C. The lysates were analyzed on a 0.8% agarose gel to score for colonies that contain plasmids larger than the empty vector (pGL3-U6-sgRNA-PGK-puromycin). Positive clones were then selected for colony PCR in a 20 μL reaction mixture that includes 200 nM forward and reverse primers (mRFPF and mRFPR), 200 μM dNTPs, and 1× OneTaq master mix (NEB), using parameters as described (Table 4).

Table 4: Colony PCR parameters.
To generate pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR, the mRFP-T2A insert and the linearized pGL3-U6-sgRNA-PGK-puromycin vector (described above) were recombined using a cold fusion cloning kit (System Biosciences, USA) by following the manufacturer’s instructions. In brief, a 5 μL sample of 50 ng vector, 100 ng insert, and 1 μL of cold fusion master mix was added to 50 μL of competent DH5α cells (System Biosciences). Following 20 min incubation on ice, the mixture was heat shocked at 42 °C for 50 sec before 250 μL of LB broth was added. The mixture was then transferred onto LB agar plate containing 100 μg/mL ampicillin for overnight incubation at 37 °C.
sgRNA vector pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR was digested with BsaI-HF (NEB) to generate 5′ overhangs. In brief, a 10 μL reaction mixture including 4 μg of the vector DNA, 1× cut-smart buffer (NEB), and 80 units of BsaI-HF was incubated at 37 °C for 1.5 h. After heat inactivation at 80 °C for 20 min, 1 unit of rSAP (NEB) was added to dephosphorylate the digested/linearized vector at 37 °C for 1.5 h. rSAP was then inactivated via a 5 min incubation at 65 °C. The linearized and dephosphorylated vector was gel purified using QIAquick gel extraction kit (Qiagen) by following the manufacturer’s instructions.
Meanwhile, sgRNA sequence targeting a glutamine codon (CAG) in exon 2 (position 45583-85) of the Munc13-1 gene was identified using the Github database (https://www.ciccialab-database.com/istop) created by Billon et al. [10]. Ten μM forward (sgRNAF) and reverse (sgRNAR) oligos containing BsaI overhangs (boldened in Table 1) were phosphorylated (37 °C × 1 h) by 200 units of T4 polynucleotide kinase (NEB) in a 10 μL reaction mixture that also contained 1× T4 ligation buffer (NEB). The oligos were annealed by incubating the mixture at 95 °C for 5 min (following a 1 h preincubation at 37 °C) and then at RT for 1 h.
Ligation was conducted in a 10 μL reaction mixture containing 100 ng digested vector, 0.1 μM annealed sgRNA oligos, 1× T4 ligase buffer, and 0.5 unit T4 ligase (NEB). Following 1 h incubation at RT, the mixture was then incubated at 65 °C for 10 min to inactivate the T4 ligase, and 5 μL of the ligation mixture was used to transform 50 μL competent DH5α cells (prepared using CaCl2 as described in [15]).
RBL-2H3 cells (ATCC, USA) were maintained in DMEM medium (high glucose; Gibco) containing 10% fetal bovine serum (FBS; Gibco) and 1× Gibco GlutaMax supplement (aka complete medium) at 5% CO2 and 37 °C in a humidified incubator. One million RBL-2H3 cells were transfected with 3 μg of pGL3-U6-sgRNAMunc13-1-PGK-mRFP-T2A-PuroR and 7 μg of pCMV BE4max using SF cell line 4D-NucleofectorTM X Kit (Lonza, Switzerland) by following the manufacturer’s instructions. Cells were then incubated in complete medium for 48 h before the addition of 2.5 μg/mL puromycin. Fluorescence microscopy was conducted before and after puromycin treatment using Leica fluorescence microscope. Single cell dilution was performed after puromycin treatment to obtain independent clones.
To screen for Munc13-1null RBL-2H3 clones, each candidate clone was grown to around 85% confluency in a 24-well plate. After the medium was removed, cells were suspended with 125 μL of 0.25% Trypsin/EDTA (Gibco, USA) for 4 min before the addition of 250 μL of complete medium. Following centrifugation at 300 g for 5 min, cell pellets were resuspended in 100 μL of PBS. After another round of centrifugation at 300 g for 5 min, cells were resuspended in 50 μL of lysis solution (10 mM Tris-Cl, pH 8.5, 25 mM KCl, 10% Triton X-100, and 0.4 μg/μL proteinase K). To prepare for RFLP (restriction fragment length polymorphism) analysis, a 799 bp fragment spanning the sgRNA target site was amplified in a 25 μL PCR (Table 5) that contained 2 μL of cell lysate (from
above), 200 nM forward and reverse primers (RFLPtempF and RFLPtempR; Table 1), 200 μM dNTPs, and 1× OneTaq master mix (NEB), using parameters as described (Table 5). PCR products were purified with Wizard PCR preps DNA purification system (Promega, USA). Approximately 120 ng
of PCR products were digested with 20 units of NheI-HF (NEB) at 37 °C for 1 h, in a 20 μL reaction mixture that also contains 1× cut-smart buffer. The reaction mixture was then incubated at 80 °C for 20 min to inactivate NheI-HF, and analyzed using (1.5%) agarose gel electrophoresis.

Table 5: PCR parameters for RFLP analysis.
To create an sgRNA vector that carries both a fluorescent marker and a puromycin-resistance marker, we decided to insert the cDNA for mRFP in between the PGK promoter and the puromycin resistance gene of the pGL3-U6-sgRNA-PGK-puromycin vector (Figure 1(a)). To accomplish this, we first used PCR to amplify the cDNA for mRFP and a downstream T2A (a self-cleaving 2A peptide) from plasmid MCS1-EF1α-RFP-T2A-Puro-pA-MCS2. T2A was included in the design to ensure that from the new sgRNA vector (pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR), mRFP, and the downstream puromycin-N-acetyltransferase (which confers puromycin resistance) are uncoupled and independently folded upon translation. The PCR product was analyzed on agarose gel electrophoresis, which revealed a DNA band that matched the predicted size of the insert (762) (Figure 2(a); lane 1). This band was therefore excised
and DNA there within was extracted. Meanwhile, pGL3-U6-sgRNA-PGK-puromycin, a commonly used sgRNA vector in base editing, was linearized by PCR at position 1168/9, which is situated downstream of the PGK promoter but immediately before the puromycin-N-acetyltransferase start codon. Judging from Figure 2(b) (compare lanes 1 and 2), the linearized vector DNA is much more abundant than the circular, template DNA, which had been methylated before its isolation from DH5α. To minimize false positives, circular template DNA remaining at the end of PCR linearization was removed via DpnI (Figure 2(b), compare lanes 2 and 3), which specifically degrade methylated DNA.
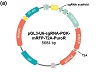
Figure 1: New sgRNA vector pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR. (a) Schematic representation of the new plasmid. (b) DNA sequence of the region between the PGK promoter and the puromycin resistance gene. The mRFP cDNA (without the stop codon) is in frame with the T2A linker and the puromycin resistance gene. The entire construct has been sequence verified. The dotted lines represent codons not depicted in the diagram.
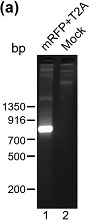
Figure 2: Cloning of pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR. (a) The mRFP-T2A insert was amplified from MCS1-EF1α-RFP-T2A-Puro-pA-MCS2 via a touchdown PCR and analyzed by agarose (2%) gel electrophoresis (lane 1). A mock control (without the DNA polymerase) was conducted in parallel (lane 2). (b) The methylated pGL3-U6-sgRNA-PGK-puromycin vector DNA was linearized via PCR, and subject to agarose (0.7%) gel electrophoresis before and after DPN1 digestion (lanes 1 and 3 resp.). The mock control for PCR lacked DNA polymerase (lane 2). (c) Colony PCR and subsequent agarose (2%) gel electrophoresis were performed to identify clones with the mRFP-T2A insert. Clone #2 was selected for DNA sequencing analysis.
Because the DNA primers used to amplify “mRFP+T2A” insert have 15 bp nucleotides at the 5′ end (Table 1) that match the DNA sequences flanking the cloning site (1168/9), the insert was cloned into the linearized vector via homology-based recombination. The recombination reaction mixture was used to transform competent DH5α, which gave rise to 13 independent colonies. Plasmids were released from these colonies via DNA cracking and one of them demonstrated slower mobility than the pGL3-U6-sgRNA-PGK-puromycin vector based on agarose gel electrophoresis. This colony was then selected for colony PCR using the same set of primers for amplifying the mRFP-T2A insert, and, according to subsequent agarose gel electrophoresis, it seems to contain the desired insert (Figure 2(c), compare lanes 3 and 1). Subsequent DNA sequencing indicated that mRFP has been inserted at the expected site, in frame with the downstream puromycin resistance marker (Figure 1(b)).
To test the efficacy of the newly generated sgRNA vector (pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR) in base editing, we decided to use it to introduce an early stop codon in Munc13-1 in RBL-2H3 cells. We started by cloning Munc13-1-specific sgRNA into pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR using the BsaI restriction site. RBL-2H3 cells were then co-transfected with pGL3-U6-sgRNAMunc13-1-PGK-mRFP-T2A-PuroR and pCMV_BE4max. Transfection efficiency was estimated to be at 62% (207 fluorescent cells over a total of 334 cells counted) based on the expression of the mRFP at 48 h time point. Puromycin was then added at 2.5 μg/mL. After an additional three days of incubation, 99% of the remaining cells (350 fluorescent cells over a total of 354 cells counted) were found positive for mRFP (Figure 3), indicating that both mRFP and puromycin-N-acetyltransferase were expressed and functional under our experimental conditions. We then went ahead to isolate the clones from transfected cells via single-cell dilution. Because the successful editing of the targeted glutamine codon (CAG) in exon 2 of rat Munc13-1 gene will introduce an NheI restriction site (Figure 4(a)), we performed NheI-based RFLP analysis on 78 clones. Sixty four of these clones (82%) exhibit patterns of monoallelic editing (Figure 4(b)), which suggest that this new system we used for genome editing is highly effective. Importantly, Clone BL7 was further analyzed via DNA sequencing and the change of CAG to TAG was confirmed (Figure 5; arrow).

Figure 3: Use of pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR in base editing. RBL-2H3 cells were transfected with pGL3-U6-sgRNAMunc13-1-PGK-mRFP-T2A-PuroR and pCMV_BE4max, and grown in complete media with puromycin. Fluorescence microscopy shows that virtually all puromycin resistant cells expressed red fluorescent signals (a) and (b). In contrast, RBL-2H3 cells that lacked pGL3-U6-sgRNAMunc13-1-PGK-mRFP-T2A-PuroR did not display red fluorescent signals (c) and (d). Scale bar, 100 μm.
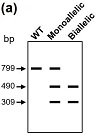
Figure 4: RFLP analysis of Munc13-1null clones. (a) Schematic diagram of NheI-based RFLP analysis of potential Munc13-1null clones. (b) A 799 bp Munc13-1 DNA fragment containing the editing site was amplified from individual clones using One-Taq DNA polymerase. Following NheI digestion and subsequent agarose (1.5%) gel electrophoresis, monoallelic mutants were distinguished from the WT.

Figure 5: DNA sequencing analysis of Clone BL7. The 799 bp Munc13-1 DNA fragments amplified from BL7 and the WT control (
Figure 4) were sequenced, revealing the base substitution in BL7 that changed Munc13-1 Gln42 into a stop codon (pointed by the red arrow).
CRISPR/Cas9-mediated base editing has become a promising approach to correct undesirable traits or diseases that are caused by single nucleotide polymorphism [16]. Although these kinds of corrections can be achieved via other CRISPR-based approaches, base editing offers two advantages: ease of use and higher efficiency. This process has been relying on sgRNA vectors that carry either a fluorescent marker or an antibiotic marker. For laboratories that have access to an FACS, a fluorescent marker would significantly simplify the screening process while enhancing the rate of success. However, for laboratories that do not have access to an FACS, it is desirable to have a more versatile sgRNA vector that expresses both a fluorescent marker and an antibiotic resistance marker. Such an sgRNA vector would allow users to enrich transfected cells and to monitor the transfection efficiency. For this very purpose, we have created pGL3-U6-sgRNA-PGK-mRFP-T2A-PuroR. It was successfully used to introduce an early nonsense mutation in Munc13-1 in RBL-2H3 cells. Based on our estimation, approximately 82% of the clones will bear a monoallelic mutation under our experimental conditions.
While we did not immediately obtain a biallelic mutant, we could accomplish this by another round of transfection using the monoallelic mutant that has been sequence verified (i.e., Clone BL7). It took us approximately 5 weeks to complete one round of editing to obtain the monoallelic mutant so we estimate it would take 5 to 10 weeks to get a biallelic mutant by following the same approach. However, alternative strategies could be designed to further improve the efficiency. For instance, two consecutive rounds of transfection could be conducted before single cell dilution and RFLP analysis. Additionally, histone deacetylase inhibitors (e.g., Romidepsin) could be used to facilitate euchromatin state, enhancing the access of Cas9 to its target site [8,17].
In conclusion, by creating a modified sgRNA vector that contains both fluorescent and antibiotic resistance markers, we have made the CRISPR/Cas9-dependent base editing system more versatile. This new sgRNA vector will be submitted to Addgene to make it widely available.
Research supported by the Mississippi INBRE, which was funded by an Institutional Development Award from the National Institute of General Medical Sciences under grant no. P20GM103476; Research of the second author supported by the National Institute of Allergy and Infectious Diseases grants R15AI133430 and R03AI169047.
The authors declare that they have no conflict of interest.
- L. Cong, F. A. Ran, D. Cox, et al., Multiplex genome engineering
using CRISPR/Cas systems, Science, 339 (2013), 819–823.
- P.Mali, L. Yang, K. M. Esvelt, et al., RNA-guided human genome
engineering via Cas9, Science, 339 (2013), 823–826.
- M. Bibikova, D. Carroll, D. J. Segal, J. K. Trautman, J. Smith, Y. G. Kim, et al., Stimulation of homologous recombination through targeted cleavage by chimeric nucleases, Mol Cell Biol, 21 (2001), 289–297.
- M. Bibikova, M. Golic, K. G. Golic, and D. Carroll, Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases, Genetics, 161 (2002), 1169–1175.
- R. Ceccaldi, B. Rondinelli, and A. D. D’Andrea, Repair pathway choices and consequences at the double-strand break, Trends Cell Biol, 26 (2016), 52–64.
- X. Li and W. D. Heyer, Homologous recombination in DNA repair and DNA damage tolerance, Cell Res, 18 (2008), 99–113.
- M. R. Lieber, The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway, Annu Rev Biochem, 79 (2010), 181–211.
- A. J. Aguirre, R. M. Meyers, B. A. Weir, F. Vazquez, C. Z. Zhang, U. Ben-David, et al., Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting, Cancer Discov, 6 (2016), 914–929.
- A. C. Komor, Y. B. Kim, M. S. Packer, J. A. Zuris, and D. R. Liu, Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage, Nature, 533 (2016), 420–424.
- P. Billon, E. E. Bryant, S. A. Joseph, et al., CRISPR-mediated
base editing enables efficient disruption of eukaryotic genes through induction of STOP codons, Mol Cell, 67 (2017), 1068–1079.e4.
- C. Kuscu, M. Parlak, T. Tufan, et al., CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations, Nat Methods, 14 (2017), 710–712.
- L. W. Koblan, J. L. Doman, C. Wilson, et al., Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction, Nat Biotechnol, 36 (2018), 843–846.
- N. Merienne, G. Vachey, L. de Longprez, et al., The self-inactivating KamiCas9 system for the editing of CNS disease genes, Cell Rep, 20 (2017), 2980–2991.
- B. Shen, W. Zhang, J. Zhang, et al., Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects, Nat Methods, 11 (2014), 399–402.
- T. Maniatis, E. F. Fritsch, and J. F. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1982.
- M. J. Landrum, J. M. Lee, M. Benson, et al., Clinvar: public archive of interpretations of clinically relevant variants, Nucleic Acids Res, 44 (2016), D862–D868.
- B. Liu, S. Chen, A. Rose, et al., Inhibition of histone deacetylase 1 (HDAC1) and HDAC2 enhances CRISPR/Cas9 genome editing, Nucleic Acids Res, 48 (2020), 517–532.
Copyright © 2022 Tolulope E. Ayo and Hao Xu. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.