Received 7 August 2024; Revised 4 November 2024; Accepted 17 November 2024; Published 21 December 2024
Reda M. Abdelhameed, Samir M. El-Medani, Fatma M. Elantabli, and Mahmoud El-Shahat, Uses of Palladium Complexes in Accelerating Chemical Reactions under Visible Light Irradiation, Journal of Transition Metal Complexes, 7 (2024), art246171. doi:10.32371/jtmc/246171
The formation of carbon-heteroatom and carbon-carbon bond structures could be catalyzed by palladium. This is one of the most powerful ways of motivation. Several investigations are
concentrating on implementing and expanding the ground-state reactivity of palladium catalysts. Recently, the catalysis of palladium induced by visible light has been studied so that transformations can be made in novel and unprecedented ways via the photo-excited state of the palladium complex, which could not be carried out under thermal circumstances. This distinguished highlight review explores the latest advances in the synthetic method of C−C bond under the catalysis of palladium in the presence of light. The present review showed desaturation reaction, Heck reactions, and Suzuki–Miyaura coupling catalyzed by palladium.
The usage of transition metal complexes as catalysts in organic synthesis has encouraged and enhanced the expansion of proceeding synthetic methodologies [1]. Currently, transition metal-catalyzed organic reactions play important parts in many areas, such as the manufacture of drugs, materials evolution, and biological sciences. Although palladium catalysis could be used both in industry and academia, most investigations have mainly concentrated on the implementation and growth of the ground-state reactivity of palladium catalysts, exceptionally for the reactions of C(sp2)-based electrophiles via traditional oxidative addition with electron pairs transfer [2].
Visible light-based organic reactions have made significant advances in response to topical interests in environmentally friendly green chemical synthesis. Transformations using readily available, cheap visible light sources are at the precedence of organic chemistry as powerful planning for the activation energy of small molecules. In this review, we focus on recent improvements that depend on visible light-based organic reactions, inclusive aerobic oxidation, evolution of hydrogen reactions, energy transfer reactions, and asymmetric reactions. This review aims to be used as a promising strategic direction for developing practical and scalable industrial processes with significant environmental benefits [3].
The catalysis of palladium by using a photo-excited way was discussed in the present review. Palladium might serve two purposes in this method: it could collect visible light and form the catalyst-metal bond by covalent forces in catalytic cycles to initiate or facilitate the creation of chemical bonds. Visible light may influence several essential primary steps in organometallic chemistry, including dissociation of ligands, reduction elimination, trans-metalation, oxidative addition, and migratory insertion/de-insertion. These affront primary procedures under thermal circumstances can be progressed rather easily with a visible light. In this study, it is possible now to make a summary of the latest recent developments that rely mainly on palladium stimulation with visible light. The use of visible light as a source of energy in palladium catalysis of organic synthesis is prospective to supply modern chances to discover modern reactions and also to resolve maintaining the present challenges that remain in palladium thermal catalysis.
Regarding the desaturation reaction, the resulting palladium can be catalyzed by visible light. It not only allows the immediate desaturation of electrons through SET activation of the photocatalytic inner field and thus eliminates β-H, but it also permits the desaturation of C−H bonds through the transport process of the hydrogen atom (Figure 1).
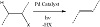
Figure 1: Desaturation reactions using Pd as catalyst.
Given the ease of access to a wide range of carboxylic acids and the synthetic adaptability of alkenes, the decarboxyolefination of alkane carboxylates to generate alkenes has substantial synthetic significance. Palladium can act as a catalyst in the presence of irradiation with (440 nm) blue LEDs, resulting in catalyzing the decarboxylative desaturation of various aliphatic carboxylates and the formation of enol ethers, aliphatic alkenes, styrenes, peptide enamides, enamides, etc., under moderate conditions. The key to successfully performing this reaction is the choice of a dual phosphine ligand system. The Pd-catalyzed decarboxylative desaturation can be used to realize a three-step divergent synthesis of Chondriamide A and Chondriamide C in overall 68% yield from simple starting materials. Mechanistic studies proposed that separately from palladium catalysis under thermal conditions, irradiation-induced palladium catalysis includes irradiation-induced single-electron transfer and dynamic ligand-dissociation/association procedures to permit two phosphine ligands to act synergistically [4]. Under mild conditions, it has been possible to remotely desaturate O-acyl hydroxamides to unsaturated amides by photoinduced and palladium-catalyzed techniques. For alkanes to be desaturated effectively and selectively, Pd(I) species and alkyl radicals must recombine to generate the alkyl Pd(II) intermediate (Figure 2). This reaction induces moderate to excellent yields of different unsaturated amides with strong selectivity at the site and no need for a terminal oxidant. This method remarkably permits the desaturation of complicated and physiologically significant compounds at a late stage [5].

Figure 2: O-acyl hydroxamides desaturation to unsaturated amides using palladium.
Figure 3 reports a photoinduced desaturation process using a palladium catalyst that may effectively transform linear amides into their α,β-unsaturated counterparts. This is a straightforward one-step reaction that can be performed at room temperature without the need for strong base/acid, sulfur/selenium, or oxidant reagents. Radical capture, radical clock, deuterium labelling experiments, and kinetic investigations have all been used to study the reaction mechanism. A radical route including aryl/alkyl Pd-radical intermediates was postulated by mechanistic studies [6].

Figure 3: Making the α,β-unsaturated compounds from the linear amides by employing Pd.
Heck reaction has been specified as the initial type of a C−C bond-making reaction that keeps the path of a Pd(0)/Pd(II) catalytic circle, the self-catalytic circle that occurs in another Pd(0)-catalyzed cross-coupling procedure (Figure 4) [7,8,9,10,11]. This chemical method took place between an unsaturated halide and an alkene in the presence of a base and a Pd catalyst creating a substituted alkene. Richard F. Heck was favored and given the Nobel Prize in Chemistry in 2010, which he shared with Ei-ichi Negishi and Akira Suzuki, for the innovation and development of this reaction [12].
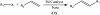
Figure 4: Heck reaction using palladium complexes.
Concerning the impact of exposure to visible light on the Heck reaction between aryl halides and styrene, it is discovered that exposure to visible light greatly increases the rate of reaction (69% versus 16%, Figure 5) [13].
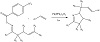
Figure 5: Palladium-catalyzed Heck reaction.
Using aromatic alkenes and blue LED irradiation at ambient temperature, a palladium-catalyzed Narasaka–Heck reaction was established (Figure 6). Since the hybrid alkyl palladium radical species could not be eliminated by the unwanted β-hydride, a variety of oxime esters were able to undergo 5-exo cyclization and then couple with olefins. Creating a nitrogen bridgehead tricycle as the fundamental framework for alkaloids demonstrated the method’s applicability [14]. A palladium-catalyzed oxidative C−H alkylation of oximes triggered by visible light has been created. This gentle procedure makes it possible to manufacture alkyl-substituted oximes with an effective C−C bond. This transformation reaction could be effectively carried out by a wide variety of primary, secondary, and tertiary alkyl bromides, iodides, and formaldoximes. Through the use of visible light, the process produces nucleophilic hybrid alkyl Pd radical intermediates that, when added to the imine moiety and then eliminated by β-hydrogen, yield substituted imines [7]. Under mild conditions, visible-light-driven, palladium-catalyzed Heck reaction of S,S-functionalized internal vinyl bromides with styrenes effectively yielded functionalized 1,3-dienes. Through a radical reaction route, this Heck reaction demonstrated tolerance of a broad range of functional groups and produced the target products in moderate to exceptional yields. It is possible to further modify the resulting diene products to produce highly functionalized trisubstituted furan derivatives [15].
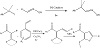
Figure 6: Oxidative C−H alkylation of oximes catalyzed by palladium.
The cross-coupling method for formalizing carbon-carbon bonds was initially disclosed by Akira Suzuki in 1979. It uses a palladium (0) complex as a catalyst to couple an organoboron molecule with an organohalide [16]. Pd-catalyzed organic synthesis by a cross-coupling technique was invented and refined by Richard F. Heck, Akira Suzuki, and Ei-ichi Negishi, who shared the 2010 Nobel Prize in Chemistry (Figure 7) [17,18]. The research details the plasmonic characteristics and catalytic efficacy of Pd anchored to TiO2 in Suzuki–Miyaura coupling (SMC) processes. In contrast to earlier studies, this semiconductor can support the production of C−C bonds for SMC since its band gap is ≥3 eV. The effective synthesis of nano Pd/TiO2 was achieved by photodeposition in the presence of sunlight. The localized surface plasmon resonance was enhanced by the size of Pd nanoparticles on the TiO2 surface. This heterogeneous photocatalyst that harvests visible light adequately encouraged substrates to couple reaction products in aqueous fluids at room temperature. Photogenerated electron-hole pairs are necessary for substrate activation under visible light irradiation [19].
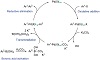
Figure 7: Suzuki reaction mechanism.
In synthetic chemistry, Suzuki coupling is an important, extensively researched, and versatile method for creating C−C bonds. However, employing palladium-based catalysts, the process has recently been thoroughly studied under thermal circumstances. With the advancement of noble-metal-free solar light-assisted for these kinds of reactions, Suzuki coupling is environmentally friendly. The photocatalytic approach is simple, efficient, and economical. It involves Suzuki-cross-coupling to form C−C bonds between aryl halides and phenylboronic acid. To do this, a cobalt(II) phthalocyanine complex is grafted onto a nickel oxide semiconductor at atmospheric pressure and room temperature (RT) (Figure 8). Several replaced aryl halides have been effectively examined using the advanced protocol and granted moderately to high yields of the required biphenyls. The prospective mechanistic pathway proposed that the conformation of photo-generated aryl radical cations from the phenylboronic acid tailed by its coupling with aryl radical anions introduced from the electron-activated aryl halides resulted in the corresponding biaryls. More importantly, the physical mixture of both components, CoPc and NiO, showed poorer capacity when compared with the sophisticated hybrid photo-catalyst. The recuperated photo-catalyst could easily be recycled for at least six runs without a considerable decrease in the activity [20].

Figure 8: Palladium-free photocatalytic Suzuki cross-coupling.
The direct catalytic C(sp2)−H alkylation of unactivated arenes with an easily accessible alkyl halide is still difficult to accomplish despite the basic significance of selective and efficient synthesis of broadly applicable alkylarenes. Unactivated arenes undergo catalytic C(sp2)−H alkylation reactions with alkyl bromides by Pd catalysis driven by visible light. Under mild circumstances, the reaction proceeds with exception of any skeletal rearrangement of the alkyl groups. Extensive mechanistic analyses utilizing computational and experimental techniques unveil a discernible Pd(0)/Pd(I) redox catalytic cycle and the source of the reactivity variations among alkyl halides [21]. Figure 9 illustrates how visible light irradiation and Pd-catalysis have been used in practice to create α-naphthyl functionalized acetates through C−C bond construction in mild reaction conditions and solvent-free environments. These reactions involve naphthylamines and α-diazo esters. It has been demonstrated that light irradiation is crucial to the reaction, most likely because it encourages the α-diazo esters to produce active carbene species [22].
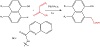
Figure 9: Pd-catalysis C−H alkylation reactions of naphthylamines and α-diazo esters.
Aryldifluoromethylated compounds are still difficult to prepare through selective functionalization of inert C(sp3)−F bonds. One such pathway involves the oxidative addition of the C(sp3)−F bond in trifluoromethylarenes (ArCF3), which is catalyzed with a transition metal (Figure 10). These precursors are readily available and reasonably priced, making them perfect for this approach. The selective defluoroarylation of trifluoromethylarenes with arylboronic acids using an unmatched excited-state palladium catalysis approach was discussed. Palladium-catalyzed visible light-induced cross-coupling allows for the transformation of several arylboronic acids and ArCF3 under mild reaction conditions. The activation of the C(sp3)−F bond in ArCF3 is reliably achieved by the oxidative addition of the C(sp3)−F bond to excited-state palladium(0) through a single electron transfer pathway, according to preliminary mechanistic research [23].

Figure 10: Transition metals catalyzed cross-coupling oxidative addition of the C(sp3)−F link in trifluoromethylarenes (ArCF3).
The explanation of a general palladium-catalyzed carbonylative process to synthesize acyl fluorides from aryl, heteroaryl, alkyl, and functionalized organic halides has been shown in Figure 11. The mechanistic analysis proposed that the reaction proceeds through the synergistic integration of visible light photoexcitation of Pd(0) to produce oxidative addition with a ligand-favored reductive elimination. These all create a unidirectional catalytic cycle that is irresponsible by the classical effect of carbon monoxide coordination. Coupling the catalytic formation of acyl fluorides with their sequently nucleophilic reactions has opened a method to carry out carbonylation reactions with unmatched enlargement, including the association of highly functionalized carbonyl-containing products [24]. The development of 1,4-dihydro-2H-3,1-benzoxazin-2-ones from 2-(1-arylvinyl)anilines has been achieved by a radical oxy-alkylation process driven by visible light and palladium, utilizing unactivated alkyl bromides and CO2. The steps in this multicomponent reaction (MCR) are as follows: (1) O2 carboxylates an amino; (2) visual light drives the reaction of alkyl bromides and Pd(0) to give Pd(I) and an alkyl radical; (3) an alkyl radical is added to the vinyl and then Pd(I) is used to oxidize it through single electron transfer (SET); and (4) cyclization takes place through intramolecular nucleophilic attack of the carboxylate anion to carbocation [25]. Oxidative carbonylation of amines to oxalamides driven by visible light with the release of hydrogen gas has been advanced. The new approach uses a simple robust Pd complex, which can even be partially recycled. A mechanistic reason is proposed and confirmed by control experiments and EPR investigations, showing that PdI formed and Pd0 was the active species. Both nitrogen and the intermediate acyl radical had been detected [26].
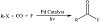
Figure 11: Synthesis of acyl fluorides using a carbonylative process catalyzed by palladium.
A decarboxylative thiocarbonylation of aryl and alkenyl sulfonium salts with oxalic acid monothioethers (OAMs) can be achieved by visible light-accelerated palladium catalysis. Sulfonium salts are exceedingly obtainable, and OAM is a readily attainable and stored reagent; this moderate reaction method can also be utilized for the synthesis of different species of thioester compounds. The reaction shows a new application of visible lightaccelerated palladium catalysis in catalytic decarboxylative cross-couplings (Figure 12) [27].
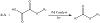
Figure 12: Decarboxylative thiocarbonylation using Pd.
The photochemical tandem cyclization/dicarbofunctionalization of unactivated alkyl halides with an alkene moiety, catalyzed by palladium, presents a promising method for generating five- or six-membered rings. By forming new C−B or C−O bonds, multisubstituted carbo- and heterocyclic compounds were created, offering a practical synthetic pathway for additional transformations. Good compatibility issue functional group, a wide substrate range, gentle conditions of reaction, and the reaction of alkene regio- and stereoselectivities are features that
distinguish this technique [28]. A simple method to obtain a wide variety of vinylpyrrolidines is to use photoinduced palladium-catalyzed annulation of 1,3-dienes with bifunctional halogenated alkylamines (Figure 13). The synthesis of complex and physiologically significant compounds as well as the diversity-oriented modifications
of the final product remarkably demonstrated the usefulness of this technique [29].

Figure 13: Photoinduced annulation of 1,3-dienes with bifunctional halogenated alkylamines catalyzed by palladium.
A recent and proper method was determined for the installation of derivatives of polyarylfuran. The coupling of allenylphosphine oxide and bromophenol or bromonaphthol could be enabled using visual light and palladium catalysis instantly supplies polyarylfuran skeletons, which involves at the same time a radical cyclization and cascade C(sp3)−P(V) bond cleavage.
This protocol faces a simple process, a wide substrate scope, and a high step economy, affording polyarylfurans in moderate to good yields (Figure 14) [30]. A combined palladium- and photoredox-catalyzed C−H olefination can be rendered to synthesize indoles product. Utilizing visual light, direct C−H activation of aromatic enamines leads to a variant of indole derivatives in good yields under simple reaction conditions [31].
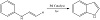
Figure 14: Combined palladium- and photoredox-catalyzed C−H olefination.
Palladium catalysis driven by visible light is reported to selectively oxy-alkylate allylamines using unactivated alkyl bromides and CO2. The only catalyst utilized in this three-component reaction is Pd(PPh3)4, which is available for purchase. Figure 15 illustrates how various alkyl bromides, including primary, secondary, and tertiary ones, undergo reactions to produce significant 2-oxazolidinones with good selectivity and yields. The easy scalability, simple derivatization of products, and mild reaction conditions offer a substantial deal of potential for use in medicinal chemistry and chemical synthesis [32].
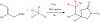
Figure 15: Selective oxy-alkylation of allylamines using Pd.
A visual-light-induced Negishi cross-coupling is authorized by the activation of a Pd0-Zn complex. By this photocatalytic method, the field of obstructed aryl halides that can be used in the Negishi coupling was safely increased. NMR spectra experiments were carried out in the presence and absence of light and guaranteed that the structure of the palladium-zinc complex is the key to speed up the oxidative addition step (Figure 16).
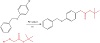
Figure 16: Visible-light-induced Negishi cross-coupling using Pd.
A modern C-1 selective mono-arylation/acylation of N-protected carbazoles with aryl diazonium salts/glyoxylic acids has been extended in visible light by a dual catalytic system involving each of Eosin Y and palladium acetate. The methodology has good functional group such as allowance and high region selectivity and providing the monosubstituted products in moderate to good yields at room temperature [33]. Directed C−H functionalization is a powerful means to functionalize otherwise unreactive C−H bonds, for which aminoquinoline amides supply a powerful and frequently used directing group. Saturated N-heterocycles are critical motifs in medicinal chemistry. However, the C−H functionalization of N-heterocycles has posed suitable large challenges, often giving incomplete conversions. On unsymmetrical substrates, poorly understood regio- and stereoselectivity considerations have inhibited more forcing conditions and consequently gave further finite yields. It has been detailed how regio- and stereo-selective C4 arylation of pyrrolidines and piperidines with C3 aminoquinoline amide directing groups is studied using both theoretical and experimental methods. Comprehensive mechanistic experiments are provided, encompassing deuteration, kinetics studies, and palladacycle isolation. Although both the trans and cis activation of the C−H bonds at C4 to the directing group happen equally, palladacycle synthesis is reversible and can start at C4. It was discussed that strain in the trans-palladacycle (ΔΔGtrans–cis∼ 6 kcal mol−1), which is preserved in subsequent transition states, is the cause of the cis-selectivity.
Hence, the oxidative addition step is stereo-determining. However, the turnover-limiting step for the catalytic cycle is reductive elimination and reduced rates and yields must pay attention to electron-poor aryl iodides. Seriously, kinetics experiments reveal a fast loss of the active Pd catalyst, likely due to the setting-up of iodide and a role for K2CO3/PivOH in catalyst turnover. Finally, the detection of an amended 4-dimethylamine-8-aminoquinoline directing group (DMAQ) was discussed. This removable auxiliary achieves >2× rate acceleration, generally amended yields, and afflicted cis-selectivity, by elevating reductive elimination. A broad reaction scope of aryl iodides is elucidated, including the late-stage functionalization of drug compounds which can be done by the use of only one equivalent of functionalized iodides [34].
Figure 17 outlines a highly modular 1,4-difunctionalization of 1,3-dienes with sulfinates/amines and bromodifluoroacetamides via a photoinduced radical relay mechanism catalyzed by palladium complex. A simple and general route to access high-value CF2-incorporated alkenes with moderate to good yields is provided by this developed protocol. The wide functional group compatibility, synthetic convenience, and easily available starting materials are excellent examples of this approach’s adaptability and flexibility [35].
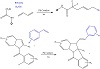
Figure 17: Synthesis of 1,4-difunctionalization of 1,3-dienes using Pd.
Moderate to good yields and good to exceptional diastereoselectivities are reported for a mild visible light-driven palladium-catalyzed radical tandem dearomatization of indoles with unactivated alkenes. The palladium complex underwent photoexcitation upon exposure to visible light, allowing it to donate a single electron to N-(2-bromobenzoyl)indoles and resulting in a hybrid palladium radical chemistry. This makes a variety of 2,3-disubstituted indoline derivatives accessible effectively and cost-effectively [36]. Figure 18 describes a one-pot, three-component alkyl-carbamoylation and cyanation of alkenes that are mildly stimulated by visible light and catalyzed by Pd. This universal transformation from easily obtained alkenes, alkyl iodides, and isocyanides enables the quick synthesis of a wide variety of useful amides and nitriles via the in-situ production of a reactive ketenimine intermediate. Using this method, an effective synthesis of amidine and tetrazole through this approach was also presented [37].
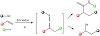
Figure 18: One-pot, three-component alkyl-carbamoylation catalyzed by Pd.
Using a unique visible-light-induced palladium-catalyzed Heck reaction for bromine sugars and aryl olefins with strong regio- and stereochemistry selectivity, the synthesis of C-glycosyl styrene is discussed. This reaction uses a straightforward and widely accessible starting material, and it occurs in a single step at room temperature. A broad variety of glycosyl bromide donors and aryl olefin substrates can be used with this technique. According to mechanistic research, a radical addition mechanism is involved [38]. It is reported that alkenes can be aerobically difunctionalized by visible light to produce isoxazolidines. Superoxides limit the radical intermediates produced when α-amino radicals from the oxidative decarboxylation of N-aryl glycine are combined with alkenes. The quick intramolecular amine oxidation of the peroxides produces valuable isoxazolidines. Zinc can easily reduce the isoxazolidine ring in acetic acid, resulting in γ-lactams with a high yield [39].
The authors declare that they have no conflict of interest.
Copyright © 2024 Reda M. Abdelhameed et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.