Departamento de Química, Universidade Estadual de Maringá, Av. Colombo, 5790, 87020-900 Maringá, PR, Brazil
Received 18 November 2021; Revised 18 January 2022; Accepted 20 January 2022; Published 22 February 2022
Angélica M. Lazarin, Marco A. G. Benetti, Denise I. Soares, Giovanna G. Alves, and Eduardo D. A. Pinto, Construction of a New Sensor with Copper(II) Complex Anchored in Calcium Phosphate Intercalated with m-Aminobenzoic Acid and Their Application in Determination of Dopamine, Journal of Transition Metal Complexes, 5 (2022), art246139. doi:10.32371/jtmc/246139
Crystalline lamellar calcium phosphate retained m-aminobenzoic acid inside its cavity without leaching. The intense infrared bands in the 1,033 cm−1 and 1,010 cm−1 intervals confirmed the presence of the phosphate groups attached to the inorganic layer, with sharp and intense peaks in X-ray diffraction patterns, which gave basal distances of 535 pm and 947 pm for the original and the intercalated compounds, respectively. The adsorption isotherm of copper(II) on a calcium phosphate host intercalated with m-aminobenzoic acid was studied in ethanol solution to give a maximum adsorption capacity of 1.43 mmoL g−1. The material was incorporated into carbon paste electrode and its electrochemical properties were investigated. However, for a dopamine solution, an enhancement of the anodic peak current was detected due to the electrocatalytic oxidation. The electrode presented the same response for at least 150 successive measurements, with a good repeatability. The modified electrode is very stable and reproducible. The sensor was applied for dopamine determination in pharmaceutical preparation (one commercial sample (Revivan)) with success.
calcium phosphate; intercalation; cyclic voltammetry; electrocatalytic oxidation; copper(II); dopamine
Phosphates intercalated with organic compounds have aroused interest, since they can act as binders for the coordination of metal ions on their surface and be used in the construction of chemically modified electrodes (CMEs), together with a carbon paste electrode (CPE), in the study of the electrochemical behavior of these compounds, in electroanalysis, and in electrocatalysis [1,2,3,4,5,6,7,8].
The use of cyclic voltammetry and/or differential pulse voltammetry, with modified chemically electrodes as the working electrode, shall be an efficient and very sensitive analytical method for studying the electrocatalytic oxidation of reagents of biological interest, such as ascorbic acid (AA), dopamine (DA), and vitamin B6 [9,10].
The present investigation deals with the electrochemical behavior of copper(II) adsorbed on calcium phosphate intercalated with m-aminobenzoic acid. This newly synthesized compound was first used to prepare a CPE and the resulting material was tested for DA oxidation. DA (3,4-dihydroxyphenylethylamine) is a catecholamine of great clinical and pharmaceutical interest, which acts as a chemical mediator in central nervous, cardiovascular, and hormonal systems; its concentration in extracellular fluid is lower than the AA present. DA deficiency in central nervous system causes serious diseases like Parkinson and Schizophrenia [11]. Therefore, its determination in vitro and in vivo has attracted a lot of attention [12,13]. The greatest interest in development of these voltammetric sensors is the advantage to be used in a living organism to detect neurotransmitters in extracellular fluid of central nervous system with the detection of DA in the presence of high concentration of AA, which is oxidized at the same potential of DA in unmodified electrodes [14,15]. An electrochemical procedure, using an innovative, stable innovator inorganic support, is now proposed.
Calcium phosphate was synthesized by slowly adding a 0.50 mol dm−3 aqueous solution of CaCl2·2H2O to a 1.50 mol dm−3 solution of dibasic ammonium phosphate and heated to 363 K. The suspension formed remained reacting for 1 h and then the precipitate formed was filtered and dried at 323 K. Then this compound was heated to 433 K, for 48 h, to eliminate ammonia, whose reactions can be written as
CaCl2·2H2O + 2(NH4)2HPO4 ⟶ (NH4)2Ca(HPO4)2·H2O + 2NH4Cl + H2O, (1)
(NH4)2Ca(HPO4)2·H2O ⟶ Ca(H2PO4)2 + 2NH3 + H2O. (2)
Approximately, 5.0 g of calcium phosphate in a conical flask with 10.0 cm3 of 1.0 mol dm−3 ethanolic solution of m-aminobenzoic acid were stirred in a thermostated bath at 298 ± 1 K and 308 ± 1 K for 12 h. The supernatant was discarded and the solid was oven dried at 323 K. This material was used to construct a CME.
For the elemental analysis of calcium and phosphorus, samples in triplicates of approximately 50 mg of calcium phosphate were placed in ostefron decomposition flasks for microwaves with a capacity of 100 cm3, for acid digestion, in the presence of 2.0 cm3 of nitric acid (65%), 2.0 cm3 of hydrofluoric acid (48%), and 1.0 cm3 of hydrogen peroxide (30%). Then these samples were taken to a Provecto analytical microwave oven, model DGT 100 plus, for mineralization of the compounds. The total time used was 18 min. Then waiting about 1 h to open the ostefron decomposition vials, shortly thereafter the hydrofluoric acid was eliminated from the samples in the presence of approximately 2 g of boric acid. Three samples of each compound were diluted in 250 cm3 flask and subsequently analyzed. Each sample was measured three times using PerkinElmer Optima 3000DV inductively coupled plasma (ICP-AES), as it has good multi-element detection sensitivity and minimal interference in the analyzed sample. The precision between replicates was less than 5% [16].
The amount of m-aminobenzoic acid intercalated into calcium phosphate was determined by nitrogen elemental analysis on a PerkinElmer 2400 Series II CHNS/O Elemental Analyzer.
X-ray diffraction patterns were obtained with nickel-filtered CuKα (0.154 nm) radiation in the interval of 2° to 65° at a speed of 0.033° s−1 and a step of 0.050° on a Shimadzu XD3-A diffractometer (30/20 kV/mA).
Infrared spectra of the samples were performed on a PerkinElmer FTIR spectrophotometer, model 1600, by using pressed KBr pellets in the 4,000–400 cm−1 range with 4 cm−1 of resolution.
The surface area measurements were performed on a Micromeritics FlowSorb 2300 instrument through gaseous nitrogen adsorption at 77 K, by applying the BET method [17].
Using the batchwise method, the adsorption isotherm for CuCl2 in ethanol solution was obtained. For each isotherm, a series of samples containing 100 mg of CaP intercalated with m-aminobenzoic acid was shaken for 4 h, as previously established [18], in an orbital bath with variable concentrations of the metal halide at a constant temperature of 298 ± 1 K. At different time intervals, an aliquot of the supernatant solution was separated and the metal ion was analyzed by complexiometric titration using EDTA as the titrant [19]. The amount of copper(II) adsorbed, nf, was determined by applying the equation nf = (ni − ns)/m, where m is the mass of the adsorbent and ni and ns are the initial and the equilibrium amounts of the number of moles of copper cations in the solution phase, respectively. The isotherms used here are those related to the Langmuir model, which was originally derived for gas adsorption on planar surfaces such as glass, mica, and platinum. The process was successfully extended to heavy metal ion adsorptions on porous surfaces [20].
The modified CPE was prepared by mixing 30 mg of the intercalated-adsorbed copper(II) calcium phosphate, 30 mg of graphite (Fluka), and a drop of mineral oil (2.0×10−2 cm3). The paste was deposited into a cavity on the surface of a platinum disk fused at the end of a glass tube with 3 mm inner diameter. This proportion was used due to a good response obtained with a preliminary test, after a detailed study of the paste composition. This electrode is referred hereafter as CaP/MABA/Cu(II).
Electrochemical measurements were performed using this CPE as the working electrode, saturated calomel electrode (SCE) as reference electrode, and a platinum wire as auxiliary electrode. The electrochemical copper(II) properties adsorbed on calcium phosphate intercalated with m-aminobenzoic acid were explored by means of cyclic voltammetry with a PAR 273A (EG&G) potentiostat-galvanostat. Experiments were performed in 0.10 mol dm−3 of phosphate buffer electrolyte solution under a pure nitrogen atmosphere and pH = 7.2 were adjusted by adding hydrochloric acid or sodium hydroxide solutions. The DA (Aldrich) was used in all experiments. The determination of DA in commercial samples (Revivan) consisted in dissolving 2.0 cm3 of Revivan in doubly distilled, degassed water and completed to 10.0 cm3.
The elemental analyses of calcium and phosphorus for the synthesized calcium phosphate compound were 26.3% and 17.2%, respectively. These values are very close to those calculated 26.5% and 17.1%. From these results, the molecular formula Ca(H2PO4)2 was proposed.
The amount of m-aminobenzoic acid intercalated in calcium phosphate at 298 ± 1 K was 9.07% nitrogen atoms, which corresponds to 6.48 mmoL g−1. And at a temperature of 308 ± 1 K it was 9.08% nitrogen atoms, which corresponds to 6.49 mmoL g−1. Therefore, in the intercalation process there was no influence of temperature.
The proposed chemical structure of the crystalline CaP interlayer is indicated in Figure 1.
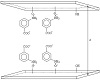
Figure 1: Representative scheme of a possible way of intercalation of MABA with the hydroxylated surface of CaP.
The layered structural feature of the crystalline calcium phosphate displays an interlayer distance which depends on cation size or molecule intercalated into the host. Thus X-ray powder diffractogram is a good tool for observing the change in interlayer distance, in order to follow the uptake process.
X-ray diffractogram provides the angle between incident and diffracted rays in the sample (2θ), showing relative intensity. The technique is not destructive and only small amounts of the powder sample are adequate to identify the crystalline phase present. Through the Bragg equation,
nλ = 2d sin θ, (3)
where λ = 1.5406 Å the wavelength of the radiation used, it can be determined by the interlamellar distance d (Å) between atomic planes of the crystalline network.
X-ray diffractograms of calcium phosphate and its form intercalated with m-aminobenzoic acid are shown in Figure 2. Calcium phosphate shows fine peaks indicating good crystallinity. The peak at 2θ = 16.73°, relative to the first reflection related to the plane (002), informs that the interlayer distance of calcium phosphate is equal to 535 pm [21]. Interlayer distance increases to 947 pm when calcium phosphate is intercalated with m-aminobenzoic acid. An increase of 412 pm compared to the original matrix shows that the intercalation occurs on the inner surface of the inorganic support (Figure 1).
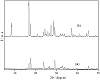
Figure 2: X-ray diffraction patterns of calcium phosphate (a) and its form intercalated (b) with m-aminobenzoic acid.
FT-IR spectra of calcium phosphate and its form intercalated with m-aminobenzoic acid are shown in Figure 3. The results show band stretching and deformation at 3,400 cm−1 and 1,550 cm−1, which is due to the OH group of the phosphate. Characteristic bands of the phosphate groups appear in 1,033 cm−1 and 1,010 cm−1 [22]. In a calcium phosphate spectrum intercalated with m-aminobenzoic acid (Figure 3(b)), it is not possible to verify the presence of bands attributed to vibrations of NH group (3,400 cm−1 to 3,300 cm−1) and other bands of inorganic matrix suffered little change with intercalation.

Figure 3: Infrared spectrum of calcium phosphate (a) and its form intercalated with m-aminobenzoic acid (b).
The area of calcium phosphate compound was equal to 20.5 ± 0.3 m2g−1 with interlayer distance of 535 pm. When this compound is intercalated with m-aminobenzoic acid, it is observed that the area increases to 21.6 ± 0.5 m2g−1. This fact may be due to amines accommodation within the
interlayer region.
Taking into account a property associated with the intercalated crystalline lamellar compound for adsorbing metal ions from ethanol solution, the corresponding isotherm for selected cations was investigated, copper(II). Initially, investigation demonstrated that the original matrix, without MABA, does not adsorb this cation. Adsorption isotherm of CaP/MABA system is shown in Figure 4. The amount of copper cation on the host surface is given by 1.43 mmoL per gram of the solid.

Figure 4: Isotherm of copper(II) complexation on the m-aminobenzoic acid in CaP compound.
After preparation of CMEs, cyclic voltammograms were obtained, in the potential range of −1.0–1.0 V, under argon. For CaP/MABA modified electrode, no peak current was observed, however, a redox coupling was verified when using CaP/MABA/Cu(II) electrode. In this case, it was observed that the average potential was Em = 0.22 V, therefore, the isotherm of copper(II) cation was investigated, which is Em = (Epc + Epa)/2, where Epa and Epc are cathodic and anodic peak potentials, respectively. In comparison with some results obtained, this wave can be attributed to the following electron transfer process [23]:
[CuII(CaP/MABA)4] + s+ + e− ⇋ [sCuI(CaP/MABA)4], (4)
where in the redox process the supporting electrolyte s+ must diffuse into or move out of the surface structured complex.
To verify the chemical stability of the copper(II) complex on calcium phosphate intercalated with m-aminobenzoic acid, several scans were made in a potential range between −1.0 V and 1.0 V versus SCE at a sweep speed 20 mVs−1 with the modified electrode immersed in a 0.10 mol dm−3 phosphate buffer solution. Results indicate that no significant variation was observed before 100 redox cycles (Figure 5). Peak current intensities remained practically constant, therefore the copper(II)
complex is strongly adhered to the calcium phosphate intercalated with m-aminobenzoic acid.

Figure 5: Plot of peak current, Ip versus number of cycles, for a CaP/MABA/Cu(II) electrode in 0.10 mol dm−3 phosphate buffer at pH 7.2.
Cyclic voltammograms reached at different scan rates indicate that the value of ΔEp (ΔEp = Epa−Epc) increases at higher ratios. Such result reflects that kinetics of electron transfer on the electrode surface is not enough fast, as a consequence of having a matrix with considerable resistance. The correlation of the peak current, Ipa and Ipc, against ν1/2 (ν is the scan rate) is linear, which is very similar to a diffusion controlled process [24]. Since the electroactive species strongly adheres to the matrix, as it was shown earlier, the mechanism may be explained by ion transport of the supporting electrolyte from electrode surface for charge compensation [25].
DA oxidation on the CaP/MABA/Cu(II) electrode surface is
shown in Figure 6. Cyclic voltammetric curves in presence of 1.3 × 10−3 mol dm−3 DA solution at pH 7.2 show important enhancement of electrode anodic peak current. The cathodic peak permanence was associated with stronger interaction between the complex of copper present in electrode and matrix surface [26]. Another explanation of cathodic peak permanence was associated with DA oxidation products. This was verified by using glassy carbon electrode in presence of DA solution. Similar behavior was also checked in others systems [27]. DA electrochemical oxidation has been studied mostly on CPEs [28,29,30] and the complete effect matches to a two-electron process, followed by two-proton process, as shown in (5); the reaction denotes the oxidation of DA into DA o-quinone (DAQ) [31].

Figure 6: Cyclic voltammogram curves for the CaP/MABA/Cu(II) electrode in the (a) absence and (b) presence of DA (0.10 mol dm−3 phosphate buffer) at pH 7.2. Scan rate of 20 mVs−1.
The effect of solution pH on the response of DA was explored (data not shown). Normally, the pH on response of DA must be taken into consideration [32]. In the present case, CaP/MABA/Cu(II) electrode was immersed in [DA] = 2.0 × 10−3 mol dm−3 and 0.10 mol dm−3 of phosphate buffer solution, and the pH was changed between 2.0 and 9.0. The current remained practically constant throughout interval, while at pH below 4.5, a considerable decrease in the response was detected. Therefore, the difference in the pH dependence of DA means that DA oxidation uses different proton and electron transfer mechanisms. According to Nernst equation, the slope of −55.7 mV/pH unit reveals that the proportion of the electron and proton involved in the reactions is 1:1. As the DA oxidation is a two-electron process, the number of protons involved is also predicted to be two [33].
The plot of current peak against DA concentration is shown in Figure 7. A linear correlation with the concentration of the acid between 0.35 × 10−3 and 1.60 × 10−3 mol dm−3 is observed with a detection limit of
0.35 × 10−3 mol dm−3. The proposed electrode presents a higher detection limit for DA which makes the sensor very attractive for determination of DA in drugs, in comparison with other modified electrodes [34,35].

Figure 7: Plot of anodic peak current (Ipa) against DA concentration in a 0.10 mol dm−3 phosphate buffer solution at pH 7.2. Scan rate of 20 mVs−1.
As stability indicator of the modified electrode towards DA, the electrode performance over 3 months period with measurements of the oxidation peak currents for 0.10 mol dm−3 DA solution in 0.35 mol dm−3 phosphate buffer solution (pH 7.2) was observed each single day. The electrode was used daily and experimental results indicated that the current responses showed a relative standard deviation of 3.50%, suggesting that the modified electrode possesses a good stability.
Major sources of interference in DA determination can be of few common coexisting species in biological fluids which could lead to either new voltammetric peaks or an overlap with the existing ones, thereby influencing their voltammetric response [36]. Citric acid, tartaric acid, glucose, uric acid, AA, and sodium chloride (NaCl) effect on voltammetric peak response of 1.5×10−3 mol dm−3 of DA was examined. It was observed that 310-fold concentrations of NaCl, tartaric acid, and glucose; 105-fold concentration of citric acid; and 11-fold concentration of uric acid and AA have no significant influence on the voltammetric peak response of 2.0 × 10−6 mol dm−3 DA. The peak current signal change was below 4% in all the cases.
The result achieved using cyclic voltammetry for a commercial sample with the CaP/MABA/Cu(II) electrode is 5.01 ± 0.12 mg cm−3 and 5.02 ± 0.18 mg cm−3, respectively, in good agreement with a confidence interval of 96% for n = 10, with the label value of 5.0 mg cm−3, thus, indicating the electrode feasibility in DA determinations in real sample. Studies have shown that the electrode is stable under operating conditions. Results obtained with this electrode are excellent, considering that other chemical elements interfere with the results. Benefits of using this electrode are mainly the ease of its preparation
and also the short response time spent on the analytical procedure. Reproducibility of the electrode was explored. Repetitive measurements were performed with DA. Results of 150 successive measurements showed a relative standard deviation of 1.03%. Therefore, modified electrodes are very stable and great reproducibility is observed.
The authors are indebted to Josué A. da Silva for manuscript revision.
The authors declare that they have no conflict of interest.
Copyright © 2022 Angélica M. Lazarin et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.